
Machine Learning Based Interatomic Potentials for Complex Thermal Transport Physics:
The high computational demand of studying thermal transport physics using first-principles methods severely limits the number of unique systems and the types of physical phenomena we can study. Machine learning (ML) based interatomic potentials have recently emerged as a promising tool for large-scale atomic simulations due to their high accuracy and low computational cost. We develop ML based interatomic potentials to predict the thermal transport properties of electronic device materials and to study complex thermal transport physics such as the effect of strain and defects on thermal transport in electronic materials.
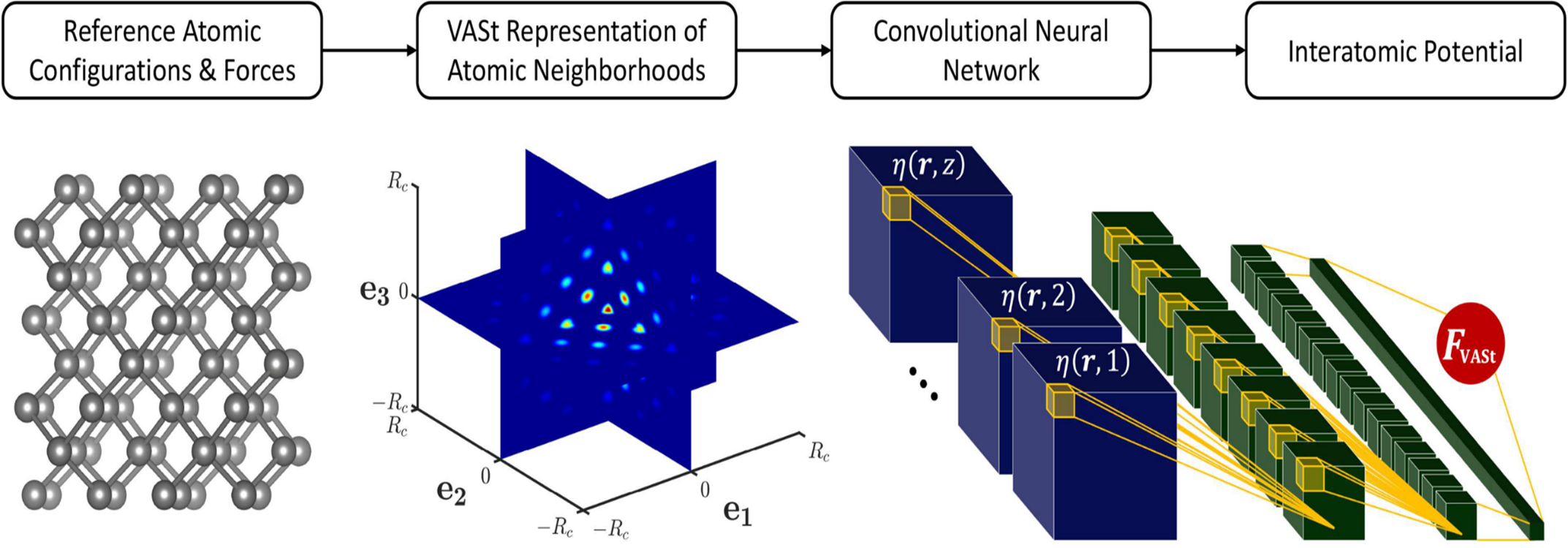
Figure 1. The framework for developing a VASt potential. (Left) Reference atomic configurations and corresponding ground-truth interatomic forces are generated for training. (Center) The reference atomic neighborhoods are mapped to their VASt representations. A 3D cross-section of a single channel of a VASt atomic neighborhood is shown. (Right) The CNN-based interatomic potential is trained. Shown, a VASt atomic neighborhood with z channels (one for each chemical element) (blue) is input to the CNN (green), which predicts the corresponding interatomic force vector F_VASt.
Phonon Transport at Material Interfaces:
Thermal transport at the interfaces of low-dimensional nanostructures such as two-dimensional (2D) molybdenum disulfide (MoS2) and graphene becomes very important in nano-electronic devices. Phonons are expected to be the dominant energy carriers for interfacial thermal transport and a fundamental understanding of phonon transport in these nanostructures and their interfaces is needed for efficient thermal management of these devices. In this work, we use first-principles density functional theory (DFT) and the atomistic Green’s function method (AGF) to study phonon transmission and thermal boundary conductance (TBC) at material interfaces. Recently, we have used these atomistic simulation methods to study (1) the relationship between interfacial electronic structure and phonon properties at the interface of monolayer MoS2 and metal substrates and (2) the effects of different lattice stacking configurations on the phonon transmission and thermal boundary conductance of hexagonal boron nitride – single layer graphene – hexagonal boron nitride (h-BN/SLG/h-BN) interfaces. The findings of this research are critical for enhancing the performance of graphene and MoS2 based electronic devices.
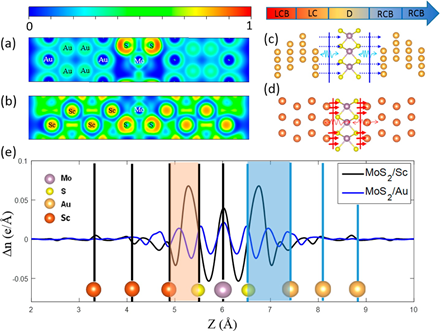
Figure 2. (a, b) Electron localization function (ELF) for the unit cell of (a) MoS2/Au and (b) MoS2/Sc. (c, d) Schematic of (c) physisorbed interface of Au/MoS2/Au and (d) chemisorbed interface of Sc/MoS2/Sc for the AGF calculations. (e) Plane-averaged electron density difference Δn (per unit cell) along out-of-plane direction showing the charge redistribution at the metal/MoS2/metal structures.
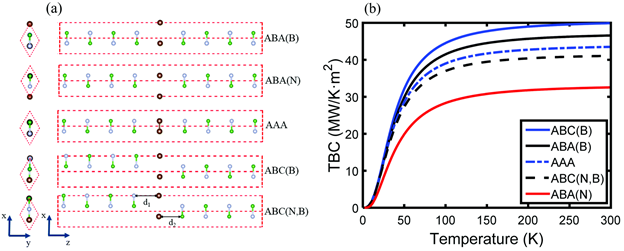
Figure 3. (a) Side views of the five lattice stacking configurations of SLG sandwiched between h-BN layers in the x–y plane and x–z plane. d1 and d2 represent the interfacial separation distances. The brown, green, and gray spheres represent carbon, boron, and nitrogen atoms, respectively. (b) Temperature dependent thermal boundary conductance at h-BN/SLG/h-BN interfaces for different lattice stacking configurations.
Phonon Transport Properties and Thermal Conductivity of Electronic Materials:
A fundamental understanding of thermal transport phenomena in electronic device materials is critical to improving thermal management of high-power dissipating electronic devices. In this work, we use first-principles density functional theory (DFT) and the Boltzmann transport equation (BTE) to study phonon transport properties such as phonon mode contributions to thermal conductivity, three-phonon scattering phase space, and phonon lifetime. In recent works, we have investigated (1) thermal transport in two-dimension electride Ca2N, (2) the effects of vacancies on the thermal conductivity of β-Ga2O3, and (3) the size-dependence and influence of vacancies and tungsten doping on phonon transport in monolayer MoSe2. The results of these studies provide important insights for the future design of electronic device materials.
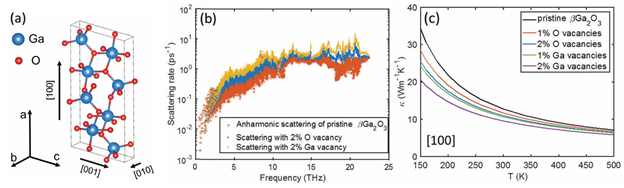
Figure 4. (a) The conventional unit cell of the bulk β-Ga2O3. (b) Angular frequency dependent anharmonic and defect-induced phonon scattering rates of the bulk β-Ga2O3 with 2% oxygen vacancies and gallium vacancies at 300 K, respectively. (c) Temperature-dependent thermal conductivity of the defective β-Ga2O3 with oxygen and gallium vacancies along the [100] direction.
Relevant Publications
1. Barry, M., Wise, K., Kalidindi, S. and Kumar, S., “Voxelized Atomic Structure Potentials: Predicting Atomic Forces with the Accuracy of Quantum Mechanics Using Convolutional Neural Networks,” The Journal of Physical Chemistry Letters, 11, 21, 9093–9099, 2020.
2. Barry, M. C., Yan, Z., Yoon, M., Kalidindi, S. R., and Kumar, S. “Phonon Transport Properties of Two-dimensional Electride Ca2N – A First-principles Study”, Applied Physics Letters, 113, 131902, 2018.
3. Yan, Z., and Kumar, S. “Phonon mode contributions to thermal conductivity of pristine and defective β-Ga2O3”, Phys. Chem. Chem. Phys., 20, 29236-29242, 2018.
4. Yan, Z., Yoon, M., and Kumar, S., “Influence of defects and doping on phonon transport properties of monolayer MoSe2”, 2D Mater, 5, 031008, 2018.
5. Yan, Z., Chen, L., Yoon, M., and Kumar, S. “The Role of Interfacial Electronic Properties on Phonon Transport in Two-Dimensional MoS2 on Metal Substrates,” ACS Applied Materials and Interfaces, 8 (48), 33299–33306, 2016.
6. Yan, Z., Chen, L., Yoon, M., Kumar, S.. “Phonon Transport at the Interfaces of Vertically Stacked Graphene and Hexagonal Boron Nitride Heterostructures.” Nanoscale, 2016, 8, 4037-4046.
7. Chen, L., Huang, Z., and Kumar, S., “Impact of Bonding at Multi-layer Graphene/metal Interfaces on Thermal Boundary Conductance.” RSC Advances, 4(68), 2014.
8. Liang Chen, Zhen Huang, and Satish Kumar. “Phonon Transmission and Thermal Conductance across Graphene/Cu Interface”. Applied Physics Letters 103(12), 4821439, 2013.
9. Liang Chen and Satish Kumar. “Thermal Transport in Graphene Supported on Copper”. Journal of Applied Physics 112(4), 043502, 2012.
Funding: NSF 