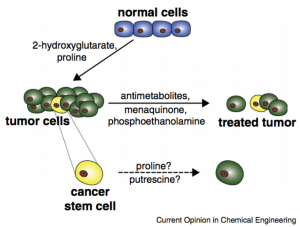
A systematic understanding of cancer metabolic pathways at the cellular level has gained importance ever since it has been reported that the glycolytic shift in aerobic metabolism, as observed by Warburg, is actually a requirement for the proliferation of cancer cells. The role played by the excessive supply of metabolites in aiding the progression or the regression of the diseased cells identified metabolites as potential candidates for the development of novel metabolism based chemotherapeutics. This work proposes to create, validate, and apply a biological data driven in silico model Computational Metabolomics (CoMet) as a tool for the prediction of metabolites that are differentially accumulated in cancer cells. The algorithm has been created using the existing and publicly-available transcriptional data to predict differences in metabolite levels in normal and cancerous cells. Measuring the changes in concentrations of metabolites in dysfunctional metabolism is a more sensitive approach than following the rates of chemical reactions directly. Systematic characterization of cancer cell metabolisms will be accomplished using two-dimensional gas chromatography coupled to mass spectrometry (GCxGC-MS), a high throughput and more sensitive analytical technique. Metabolite profiling experiments will be performed initially on Jurkat and lymphoblastic cells and later extended to other cancer cell lines to provide a broad metabolomics dataset to identify and test metabolites with anti-proliferative effects corresponding to the transcriptional dataset. This approach may help establish the idea that metabolites themselves can function as anticancer agents and subsequent experiments will be performed to identify the metabolic impacts of metabolite supplementation. This data will then be integrated into CoMet to validate and improve its accuracy of prediction of metabolites as anti-cancer agents.