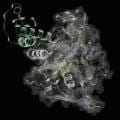
Extraterrestrial Life May Be Ubiquitous, Georgia Tech Research Suggests
Jeffrey Skolnick and coworkers at the Georgia Tech School of Biology have shown that the ability to catalyze biochemical reactions is an intrinsic property of protein molecules, defined only by their structure and the principles of chemistry and physics. Their study was published on Feb. 23, 2016, in the open-access journal F1000Research.
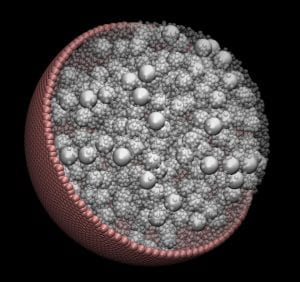
Large-Scale Modeling Shows Confinement Effects on Cell Macromolecules
Using large-scale computer modeling, researchers have shown the effects of confinement on macromolecules inside cells – and taken the first steps toward simulating a living cell, a capability that could allow them to ask “what-if” questions impossible to ask in real organisms.
Featured on NSF Science 360 NEWS and Scientific Computing
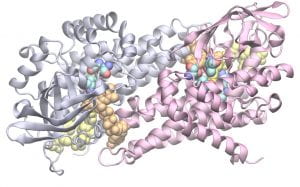
From an SNP to a Disease: A Comprehensive Statistical Analysis
Understanding and linking at the molecular level a disease phenotype to a specific genotype often requires going through a protein structure and function. In this issue of Structure, Gao et al. (2015) perform large scale analysis of available sequences and structural data for more than 6000 mutants representing more than 600 proteins to uncover some interesting structural effects of disease-associated mutations.

SURA Honors Georgia Tech Biologist, Jeffrey Skolnick As Distinguished Scientist
Washington, DC – The Southeastern Universities Research Association today announced that Jeffrey Skolnick, Director of the Integrated Biosystems Institute at Georgia Institute of Technology, will receive its 2014 SURA Distinguished Scientist Award.
The annual honor goes to a research scientist whose extraordinary work fulfills the SURA mission to “advance collaborative research and education” in the Southeast and nation. The award and its $10,000 honorarium will be presented to Dr. Skolnick on March 18, in conjunction with the SURA Board of Trustees meeting being held at the West Virginia University in Morgantown.
“SURA is proud to honor such a prolific and gifted researcher as Jeffrey Skolnick. His work in the field of protein structure and drug discovery epitomizes the importance of basic research in our society,” said Charles W. Steger, President of Virginia Tech and Chair of the SURA Council of Presidents. “As scientists like Professor Skolnick improve our knowledge of biological functions and reactions, we’ll eventually improve our quality of life and economic advancement.”
Skolnick is the author or co-author of over 350 journal articles in the fields of systems and computational biology and his cutting edge research on protein structure and function has provided remarkable insights into the relative roles of physics and evolution in dictating the properties of protein structure and function and holds the potential to dramatically accelerate and enhance the drug discovery process.
Over his career, Skolnick has made significant scientific contributions. He developed the first coarse grained model for protein structure prediction, the first successful multiscale modeling approach to structure prediction, the first effective medium model for a membrane that enabled the successful prediction of peptide orientation and conformation with respect to the membrane, Fuzzy Functional Forms that were the first low resolution approach to protein function prediction, and the highly accurate EFICAz approach to enzyme function inference. His more recent work has significant applications to both drug discovery and to improving our fundamental understanding of the possible origin of life.
Born in Brooklyn, New York, Dr. Skolnick received his bachelor’s degree in Chemistry from Washington University in St. Louis, and his Masters and Ph.D. from Yale University. Among Skolnick’s other honors and awards are: Biophysical Society Fellow (2004), Stockton Kimball Award, University at Buffalo (2003), Joseph F. Foster Lecturer, Purdue University (2001), American Association for the Advancement of Science Fellow (2001), and the Alfred P. Sloan Foundation Research Fellow (1983.)
The SURA Distinguished Scientist Award was established in 2007, commemorating the organization’s 25th Anniversary. SURA’s Development & Relations Committee manages the solicitation, screening and selection of the recipient from a SURA member institution. The president and trustee of each of SURA’s 62 member research universities is eligible to make one nomination for the Distinguished Scientist Award.
The award and honorarium will be presented to Dr. Skolnick at a reception and dinner in Morgantown on March 18.

Super PSiFR: an automated pipeline for protein structure prediction, function annotation, and virtual screening.
In the post-genomic era, the annotation of protein function facilitates the understanding of various biological processes. Over the past decade, we have developed a suite of methods for predicting protein tertiary structure and function, ligand-binding sites, protein-protein interactions, protein-DNA interactions, virtual ligand screening, drug targets of a given compound, as well as tools for the large-scale comparison of protein structures, interfaces, or pockets among all known, experimentally determined structures deposited in the Protein Data Bank. To serve the scientific community, we announce a unified, intuitive, web-based server, Super PSiFR, which combines all these tools that we have developed. In the first beta-release, Super PSiFR provides working modules for protein structure prediction, small-molecule binding prediction, DNA-binding predictions, enzymatic function predictions, and virtual screening. To try it out, simply upload a sequence, a structure, or a small-molecule compound, and select desired modules.
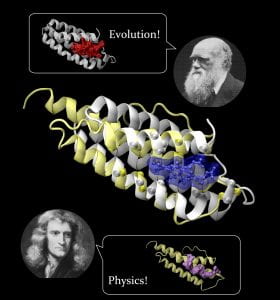
Study Suggests Drug Side Effects Inevitable; Basic Physics Enabled Early Biochemistry
A new study of both computer-created and natural proteins suggests that the number of unique pockets – sites where small molecule pharmaceutical compounds can bind to proteins – is surprisingly small, meaning drug side effects may be impossible to avoid. The study also found that the fundamental biochemical processes needed for life could have been enabled by the simple physics of protein folding.
Studying a set of artificial proteins and comparing them to natural proteins, researchers at the Georgia Institute of Technology have concluded that there may be no more than about 500 unique protein pocket configurations that serve as binding sites for small molecule ligands. Therefore, the likelihood that a molecule intended for one protein target will also bind with an unintended target is significant, said Jeffrey Skolnick, a professor in the School of Biology at Georgia Tech. Read more…

“Why not consider a spherical protein? Implications for backbone hydrogen bonding for protein structure and function.”
INSIDE COVER: Physical Chemistry Chemical Physics.
Abstract: The intrinsic ability of protein structures to exhibit the geometric features required for molecular function in the absence of evolution is examined in the context of three systems: the reference set of real, single domain protein structures, a library of computationally generated, compact homopolypeptides, artificial structures with protein-like secondary structural elements, and quasi-spherical random proteins packed at the same density as proteins but lacking backbone secondary structure and hydrogen bonding. Without any evolutionary selection, the library of artificial structures has similar backbone hydrogen bonding, global shape, surface to volume ratio and statistically significant structural matches to real protein global structures. Moreover, these artificial structures have native like ligand binding cavities, and a tiny subset has interfacial geometries consistent with native-like protein–protein interactions and DNA binding. In contrast, the quasi-spherical random proteins, being devoid of secondary structure, have a lower surface to volume ratio and lack ligand binding pockets and intermolecular interaction interfaces. Surprisingly, these quasi-spherical random proteins exhibit protein like distributions of virtual bond angles and almost all have a statistically significant structural match to real protein structures. This implies that it is local chain stiffness, even without backbone hydrogen bonding, and compactness that give rise to the likely completeness of the library solved single domain protein structures. These studies also suggest that the packing of secondary structural elements generates the requisite geometry for intermolecular binding. Thus, backbone hydrogen bonding plays an important role not only in protein structure but also in protein function. Such ability to bind biological molecules is an inherent feature of protein structure; if combined with appropriate protein sequences, it could provide the non-zero background probability for low-level function that evolution requires for selection to occur.

New Study Classifies and Analyzes Protein-Protein Interfaces
Interactions between proteins are at the heart of cellular processes, and those interactions depend on the interfaces where the direct physical contact occurs. A new study published this week suggests that there may be roughly a thousand structurally-distinct protein-protein interfaces — and that their structures depend largely on the simple physics of the proteins.

Super PSiFR: an automated pipeline for protein structure prediction, function annotation, and virtual screening.
In the post-genomic era, the annotation of protein function facilitates the understanding of various biological processes. Over the past decade, we have developed a suite of methods for predicting protein tertiary structure and function, ligand-binding sites, protein-protein interactions, protein-DNA interactions, virtual ligand screening, drug targets of a given compound, as well as tools for the large-scale comparison of protein structures, interfaces, or pockets among all known, experimentally determined structures deposited in the Protein Data Bank. To serve the scientific community, we announce a unified, intuitive, web-based server, Super PSiFR, which combines all these tools that we have developed. In the first beta-release, Super PSiFR provides working modules for protein structure prediction, small-molecule binding prediction, DNA-binding predictions, enzymatic function predictions, and virtual screening. To try it out, simply upload a sequence, a structure, or a small-molecule compound, and select desired modules.
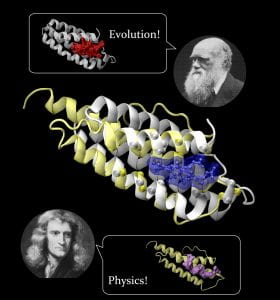
Study Suggests Drug Side Effects Inevitable; Basic Physics Enabled Early Biochemistry
A new study of both computer-created and natural proteins suggests that the number of unique pockets – sites where small molecule pharmaceutical compounds can bind to proteins – is surprisingly small, meaning drug side effects may be impossible to avoid. The study also found that the fundamental biochemical processes needed for life could have been enabled by the simple physics of protein folding.
Studying a set of artificial proteins and comparing them to natural proteins, researchers at the Georgia Institute of Technology have concluded that there may be no more than about 500 unique protein pocket configurations that serve as binding sites for small molecule ligands. Therefore, the likelihood that a molecule intended for one protein target will also bind with an unintended target is significant, said Jeffrey Skolnick, a professor in the School of Biology at Georgia Tech. Read more…

“Why not consider a spherical protein? Implications for backbone hydrogen bonding for protein structure and function.”
INSIDE COVER: Physical Chemistry Chemical Physics.
Abstract: The intrinsic ability of protein structures to exhibit the geometric features required for molecular function in the absence of evolution is examined in the context of three systems: the reference set of real, single domain protein structures, a library of computationally generated, compact homopolypeptides, artificial structures with protein-like secondary structural elements, and quasi-spherical random proteins packed at the same density as proteins but lacking backbone secondary structure and hydrogen bonding. Without any evolutionary selection, the library of artificial structures has similar backbone hydrogen bonding, global shape, surface to volume ratio and statistically significant structural matches to real protein global structures. Moreover, these artificial structures have native like ligand binding cavities, and a tiny subset has interfacial geometries consistent with native-like protein–protein interactions and DNA binding. In contrast, the quasi-spherical random proteins, being devoid of secondary structure, have a lower surface to volume ratio and lack ligand binding pockets and intermolecular interaction interfaces. Surprisingly, these quasi-spherical random proteins exhibit protein like distributions of virtual bond angles and almost all have a statistically significant structural match to real protein structures. This implies that it is local chain stiffness, even without backbone hydrogen bonding, and compactness that give rise to the likely completeness of the library solved single domain protein structures. These studies also suggest that the packing of secondary structural elements generates the requisite geometry for intermolecular binding. Thus, backbone hydrogen bonding plays an important role not only in protein structure but also in protein function. Such ability to bind biological molecules is an inherent feature of protein structure; if combined with appropriate protein sequences, it could provide the non-zero background probability for low-level function that evolution requires for selection to occur.

New Study Classifies and Analyzes Protein-Protein Interfaces
Interactions between proteins are at the heart of cellular processes, and those interactions depend on the interfaces where the direct physical contact occurs. A new study published this week suggests that there may be roughly a thousand structurally-distinct protein-protein interfaces — and that their structures depend largely on the simple physics of the proteins.

Factors Beyond Crowding
Using large-scale computer simulations, researchers at the Georgia Institute of Technology have identified the most important factors affecting how molecules move through the crowded environment inside living cells.
