Congratulations to Hargobing Singh, Ph.D. Student in Bioinformatics, as he has been awarded the Center for Programs to Increase Engagement in the Sciences (C-PIES) Research Experience Fund! This award will support his graduate research and overall career goals. The funds can be used towards travel expenses for his research experience.

COVER: Journal of Chemical Information and Modeling
Entabolons: How Metabolites Modify the Biochemical Function of Proteins and Cause the Correlated Behavior of Proteins in Pathways
Although there are over 100,000 distinct human metabolites, their biological significance is often not fully appreciated. Metabolites can reshape the protein pockets to which they bind by COLIG formation, thereby influencing enzyme kinetics and altering the monomer–multimer equilibrium in protein complexes. Binding a common metabolite to a set of protein monomers or multimers results in metabolic entanglements that couple the conformational states and functions of nonhomologous, nonphysically interacting proteins that bind the same metabolite. These shared metabolites might provide the collective behavior responsible for protein pathway formation. Proteins whose binding and functional behavior is modified by a set of metabolites are termed an “entabolon”─a portmanteau of metabolic entanglement and metabolon. 55%–60% (22%–24%) of pairs of nonenzymatic proteins that likely bind the same metabolite have a p-value that they are in the same pathway, which is <0.05 (0.0005). Interestingly, the most populated pairs of proteins common to multiple pathways bind ancient metabolites. Similarly, we suggest how metabolites can possibly activate, terminate, or preclude transcription and other nucleic acid functions and may facilitate or inhibit the binding of nucleic acids to proteins, thereby influencing transcription and translation processes. Consequently, metabolites likely play a critical role in the organization and function of biological systems.
 New AI Tool Identifies Better Antibody Therapies
New AI Tool Identifies Better Antibody Therapies
From sending cancer into remission to alleviating Covid-19 symptoms, immunotherapy can provide revolutionary disease treatments. Immunotherapies use antibodies — proteins that bind to cell markers called antigens — to target and eliminate the antigen. But despite how effective immunotherapy can be, it isn’t widely used because finding the right antibodies to develop treatments is challenging, time-consuming work.
Georgia Tech researchers are making this process a little easier, though. Their new tool, AF2Complex, used deep learning to predict which antibodies could bind to Covid-19’s infamous spike protein. The researchers created input data for the deep-learning model using sequences of known antigen binders. This method correctly predicted 90% of the best antibodies in one test with 1,000 antibodies and was recently published in Proceedings of the National Academy of Sciences. Treating Covid-19 is just the start of its potential.
 Trial Begins For ‘Probabilistic Approach’ To Diagnosing Ovarian Cancer
Trial Begins For ‘Probabilistic Approach’ To Diagnosing Ovarian Cancer
Scientists in the Georgia Tech Integrated Cancer Research Center (ICRC) are proposing a new, more realistic approach to diagnosing cancer that provides a probabilistic statement about the likelihood of developing it, much like how cholesterol tests are used to assess heart disease risk. Probabilities are based on an individual’s metabolic profile and the first population group to be targeted are women at high risk for ovarian cancer, according to John McDonald, Ph.D., professor emeritus in the school of biological sciences at Georgia Institute of Technology and founding director of the ICRC.
*Also featured on Clinical Research News.
Researchers Leverage AI to Develop Early Diagnostic Test for Ovarian Cancer
For over three decades, a highly accurate early diagnostic test for ovarian cancer has eluded physicians. Now, scientists in the Georgia Tech Integrated Cancer Research Center (ICRC) have combined machine learning with information on blood metabolites to develop a new test able to detect ovarian cancer with 93 percent accuracy among samples from the team’s study group.
*Also featured on the New York Post, Atlanta New First, Medical Xpress, News Medical Life Sciences, Medscape, AJMC, ScienceDaily, Life Technology, Omics Tutorials, Oncology News Australia, Newswise and Eurekalert.
 The University System of Georgia (USG) Board of Regents has affirmed the renewal of Jeffrey Skolnick as a Regents’ Professor!
The University System of Georgia (USG) Board of Regents has affirmed the renewal of Jeffrey Skolnick as a Regents’ Professor!
Regents’ distinctions may be granted for a period of three years by the Board of Regents (BOR) to outstanding faculty members from Georgia Tech, Augusta University, Georgia State University, the University of Georgia, and, in special circumstances, other USG institutions. A Regents’ professor, researcher, or entrepreneur distinction is awarded only after unanimous recommendation from the president of the recipient’s university, their chief academic officer and dean, as well as three additional members of the faculty who are named by the university president.

Mu Gao wins the 2023 Outstanding Sr. Research Faculty Award!
Mu Gao was awarded with the 2023 Outstanding Sr. Research Faculty Award by the Georgia Tech College of Sciences at the Spring Science Celebration, Celebrating Excellence in Research, Teaching, and Community hosted by Susan Lozier, Dean of the College of Sciences and Betsy Middleton and john Clark Sutherland Chair.
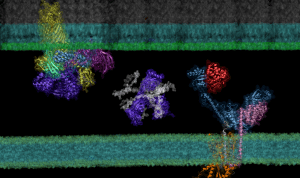 AF2Complex ‘Computational Microscope’ Predicts Protein Interactions, Potential Paths to New Antibiotics
AF2Complex ‘Computational Microscope’ Predicts Protein Interactions, Potential Paths to New Antibiotics
Though it is a cornerstone of virtually every process that occurs in living organisms, the proper folding and transport of biological proteins is a notoriously difficult and time-consuming process to experimentally study.
 Computing function from form
Computing function from form
Georgia Tech and Oak Ridge National Laboratory researchers extend AI’s prediction prowess to complex protein structures.
Over the past two years, artificial intelligence has shown it can predict what many cellular components look like. For instance, the AlphaFold deep-learning tool developed by Google sister company DeepMind has decoded how nearly every amino acid sequence folds into defined shapes.

AF2Complex: Researchers Leverage Deep Learning to Predict Physical Interactions of Protein Complexes
A computational tool developed to predict the structure of protein complexes – the molecular machinery that makes life possible – is lending new insights into the biomolecular mechanisms of their function.
Scientists use Summit supercomputer, deep learning to predict protein functions at genome scale
A team of scientists led by the Department of Energy’s Oak Ridge National Laboratory and the Georgia Institute of Technology is using supercomputing and revolutionary deep learning tools to predict the structures and roles of thousands of proteins with unknown functions.
 Georgia Tech researchers making breakthroughs treating COVID-19
Georgia Tech researchers making breakthroughs treating COVID-19
WSB-TV Channel 2 Action News coverage of MOATAI-VIR.
 Emory, Georgia Tech researchers develop AI-tool to predict COVID-19 symptoms
Emory, Georgia Tech researchers develop AI-tool to predict COVID-19 symptoms
Researchers at Emory and the Georgia Institute of Technology (Georgia Tech) published a paper on Oct. 21 detailing an artificial intelligence-based tool that is able to predict COVID-19 symptoms and suggest specific FDA-approved drugs.

AI Tool Pairs Protein Pathways with Clinical Side Effects, Patient Comorbidities to Suggest Targeted Covid-19 Treatments
The symptoms and side effects of Covid-19 are scattered across a diagnostic spectrum. Some patients are asymptomatic or experience a mild immune response, while others report significant long-term illnesses, lasting complications, or suffer fatal outcomes.
Three researchers from the Georgia Institute of Technology and one from Emory University are trying to help clinicians sort through these factors and spectrum of patient outcomes by equipping healthcare professionals with a new “decision prioritization tool.” PDF
 Jeffrey Skolnick, Andre Ghetti (ANABIOS Corporation), Nicole Jung (Karlsruhe Institute of Technology) and Hongyi Zhou win the 2020 NCATS ASPIRE Reduction-to-Practice Challenge, Stage 2, Milestone 1: Prototype Development and Milestones Delivery for their application entitled “Development of a Comprehensive Integrated Platform for Translational Innovation in Pain, Opioid Abuse Disorder and Overdose”
Jeffrey Skolnick, Andre Ghetti (ANABIOS Corporation), Nicole Jung (Karlsruhe Institute of Technology) and Hongyi Zhou win the 2020 NCATS ASPIRE Reduction-to-Practice Challenge, Stage 2, Milestone 1: Prototype Development and Milestones Delivery for their application entitled “Development of a Comprehensive Integrated Platform for Translational Innovation in Pain, Opioid Abuse Disorder and Overdose”
The Reduction-to-Practice Challenge consists of three stages: (1) Planning (winners announced in April 2021); (2) Prototype Development and Milestone Delivery; and (3) Prototype Delivery, Independent Validation and Testing. This stage of the challenge was the Prototype development.
ASPIRE-ing To Find Fast Solutions to the Opioid Health Crisis
School of Biological Sciences’ Jeffrey Skolnick and Hongyi Zhou are part of an award-winning NIH effort to create innovative, AI-powered platforms for discovering new pain management drugs — and identify immediate solutions.
 Jeffrey Skolnick, Andre Ghetti (ANABIOS Corporation), Nicole Jung (Karlsruhe Institute of Technology) and Hongyi Zhou win the 2020 NCATS ASPIRE Reduction-to-Practice Challenge, Stage 1 for their application entitled “Development of a Comprehensive Integrated Platform for Translational Innovation in Pain, Opioid Abuse Disorder and Overdose”
Jeffrey Skolnick, Andre Ghetti (ANABIOS Corporation), Nicole Jung (Karlsruhe Institute of Technology) and Hongyi Zhou win the 2020 NCATS ASPIRE Reduction-to-Practice Challenge, Stage 1 for their application entitled “Development of a Comprehensive Integrated Platform for Translational Innovation in Pain, Opioid Abuse Disorder and Overdose”
The goal of this Challenge is to combine the best solutions and develop a working platform that integrates the four component areas. The Reduction-to-Practice Challenge consists of three stages: (1) Planning; (2) Prototype Development and Milestone Delivery; and (3) Prototype Delivery, Independent Validation and Testing. The first stage of this Challenge required the submission of a plan for the reduction-to-practice of a platform that integrates the four component areas into a comprehensive solution.

MOATAI-VIR: Hot Topic of the Day on 02/02/2021 on the Centers for Disease Control and Prevention, Public Health Genomics and Precision Health Knowledge Base webpage.
Astore, C, Zhou H, Jacob J, Skolnick J. 2021. MOATAI-VIR – an AI algorithm that predicts severe adverse events and molecular features for COVID-19’s complications. medRxiv 2021.01.29.21250712; doi: https://doi.org/10.1101/2021.01.29.21250712. PDF
 Jeffrey Skolnick Receives Regents Recognition
Jeffrey Skolnick Receives Regents Recognition
Seven Georgia Tech Faculty Members Receive Regents Recognition – The University System of Georgia (USG) Board of Regents (BOR) appointed seven Georgia Tech faculty members Regents Professors, the highest academic recognition bestowed by the USG. The seven Regents Professors are:
- Marilyn Brown, Brook Byers Professor in Sustainable Manufacturing in the School of Public Policy
- Suresh Sitaraman, Morris M. Bryan Jr. Professor in Mechanical Engineering in the George W. Woodruff School of Mechanical Engineering
- Jeffrey Skolnick, Mary and Maisie Gibson Chair in Computational Systems Biology and GRA Eminent Scholar in the School of Biological Sciences
- Prasad Tetali, professor in the School of Mathematics and the School of Computer Science
- Vigor Yang, professor in the Daniel Guggenheim School of Aerospace Engineering
- Lisa Yaszek, professor in the School of Literature, Media, and Communication
- Ellen Zegura, Stephen Fleming Chair in the School of Computer Science

Origin of Life’s Handedness and Protein Biochemistry
Examine your hands. The right is a mirror image of the left. They look very similar, but you know they’re not when you try to put your left hand inside a right glove.
The molecules of life have a similar handedness. Proteins for example are like your left hand, made up of amino acids that are all left-handed. This phenomenon is called chirality. How chiral systems emerged is one of the key questions of origins-of-life research.
 On the possible origin of protein homochirality, structure, and biochemical function
On the possible origin of protein homochirality, structure, and biochemical function
Living systems contain mainly chiral macromolecules, including proteins. How L-chiral proteins emerged from demi-chiral mixtures is unknown. Our simulations show that, compared to contemporary proteins, demi-chiral proteins have shorter regular secondary structures due to fewer internal hydrogen bonds, but similar global folds and small molecule binding sites. Demi-chiral proteins contain L-chiral substructures matching native active site geometries. Among the most frequently generated enzymes with native active site residues are ancient functions associated with metabolism and replication. This suggests that demi-chiral proteins could engage in early metabolism, creating the feedback loop for transcription and cell formation partly responsible for life’s emergence.
 Jeffrey Skolnick, Mu Gao, and Hongyi Zhou win the NCATS ASPIRE Design Challenge 3: Predictive Algorithms for Translational Innovation in Pain, Opioid Use Disorder and Overdose
Jeffrey Skolnick, Mu Gao, and Hongyi Zhou win the NCATS ASPIRE Design Challenge 3: Predictive Algorithms for Translational Innovation in Pain, Opioid Use Disorder and Overdose

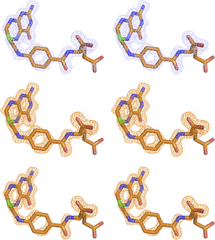 Time-resolved x-ray crystallography capture of a slow reaction tetrahydrofolate intermediate
Time-resolved x-ray crystallography capture of a slow reaction tetrahydrofolate intermediate
Structural Dynamics – Editor’s Pick
Jeffrey Skolnick: 2018 Sigma Xi Sustained Research Award – Applying computational systems biology to improve human health


Georgia Tech has named Jeffrey Skolnick the recipient of the 2018 Sigma Xi Sustained Research Award. The award recognizes Skolnick’s exceptional sustained imagination and productivity in the fields of systems biology, computational biology, bioinformatics, cancer metabolomics, protein structure prediction and evolution, drug design, and simulations of cellular processes.
Rational Design of Novel Allosteric Dihydrofolate Reductase Inhibitors Showing Antibacterial Effects on Drug-Resistant Escherichia coli Escape Variants
Highly Cited – Top Altmetric 2017 Articles
Drug Design Strategy Boosts the Odds Against Resistance Development
A new rational drug design technique that uses a powerful computer algorithm to identify molecules that target different receptor sites on key cellular proteins could provide a new weapon in the battle against antibiotic resistance, potentially tipping the odds against the bugs.
Featured on: ScienceDaily, SciCentral, Medical Web Times and The EDGE
DNA Internal Motion Likely Accelerates Protein Target Search in a Packed Nucleoid dictates to a great extent the protein motion. See the article by Chow and Skolnick in the June 6 issue of Biophysical Journal.
Related Article (news.gatech.edu): Rattling DNA Hustles Transcribers to Targets

Georgia Tech To Develop Tools To Improve Drug Efficacy
The National Institutes of Health has awarded College of Sciences’ Jeffrey Skolnick a $2.44M grant over five years. The Mary and Maisie Gibson Chaired professor in the School of Biology and a Georgia Research Alliance Eminent Scholar in Computational Systems Biology, Skolnick aims to develop tools to comprehensively annotate the parts of the human genome that are translated into proteins, known as the exome. Ultimately, the goal is to predict other uses of drugs already approved by the Federal Drug Administration (FDA) and thus accelerate the treatment of patients with intractable diseases.
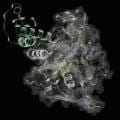
Extraterrestrial Life May Be Ubiquitous, Georgia Tech Research Suggests
Jeffrey Skolnick and coworkers at the Georgia Tech School of Biology have shown that the ability to catalyze biochemical reactions is an intrinsic property of protein molecules, defined only by their structure and the principles of chemistry and physics. Their study was published on Feb. 23, 2016, in the open-access journal F1000Research.
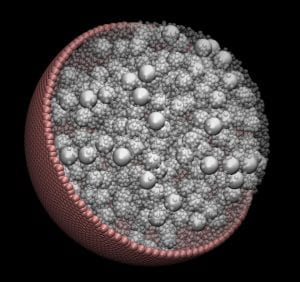
Large-Scale Modeling Shows Confinement Effects on Cell Macromolecules
Using large-scale computer modeling, researchers have shown the effects of confinement on macromolecules inside cells – and taken the first steps toward simulating a living cell, a capability that could allow them to ask “what-if” questions impossible to ask in real organisms.
Featured on NSF Science 360 NEWS and Scientific Computing
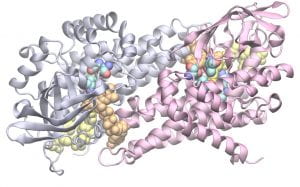
From an SNP to a Disease: A Comprehensive Statistical Analysis
Understanding and linking at the molecular level a disease phenotype to a specific genotype often requires going through a protein structure and function. In this issue of Structure, Gao et al. (2015) perform large scale analysis of available sequences and structural data for more than 6000 mutants representing more than 600 proteins to uncover some interesting structural effects of disease-associated mutations.

SURA Honors Georgia Tech Biologist, Jeffrey Skolnick As Distinguished Scientist
Washington, DC – The Southeastern Universities Research Association today announced that Jeffrey Skolnick, Director of the Integrated Biosystems Institute at Georgia Institute of Technology, will receive its 2014 SURA Distinguished Scientist Award.
The annual honor goes to a research scientist whose extraordinary work fulfills the SURA mission to “advance collaborative research and education” in the Southeast and nation. The award and its $10,000 honorarium will be presented to Dr. Skolnick on March 18, in conjunction with the SURA Board of Trustees meeting being held at the West Virginia University in Morgantown.
“SURA is proud to honor such a prolific and gifted researcher as Jeffrey Skolnick. His work in the field of protein structure and drug discovery epitomizes the importance of basic research in our society,” said Charles W. Steger, President of Virginia Tech and Chair of the SURA Council of Presidents. “As scientists like Professor Skolnick improve our knowledge of biological functions and reactions, we’ll eventually improve our quality of life and economic advancement.”
Skolnick is the author or co-author of over 350 journal articles in the fields of systems and computational biology and his cutting edge research on protein structure and function has provided remarkable insights into the relative roles of physics and evolution in dictating the properties of protein structure and function and holds the potential to dramatically accelerate and enhance the drug discovery process.
Over his career, Skolnick has made significant scientific contributions. He developed the first coarse grained model for protein structure prediction, the first successful multiscale modeling approach to structure prediction, the first effective medium model for a membrane that enabled the successful prediction of peptide orientation and conformation with respect to the membrane, Fuzzy Functional Forms that were the first low resolution approach to protein function prediction, and the highly accurate EFICAz approach to enzyme function inference. His more recent work has significant applications to both drug discovery and to improving our fundamental understanding of the possible origin of life.
Born in Brooklyn, New York, Dr. Skolnick received his bachelor’s degree in Chemistry from Washington University in St. Louis, and his Masters and Ph.D. from Yale University. Among Skolnick’s other honors and awards are: Biophysical Society Fellow (2004), Stockton Kimball Award, University at Buffalo (2003), Joseph F. Foster Lecturer, Purdue University (2001), American Association for the Advancement of Science Fellow (2001), and the Alfred P. Sloan Foundation Research Fellow (1983.)
The SURA Distinguished Scientist Award was established in 2007, commemorating the organization’s 25th Anniversary. SURA’s Development & Relations Committee manages the solicitation, screening and selection of the recipient from a SURA member institution. The president and trustee of each of SURA’s 62 member research universities is eligible to make one nomination for the Distinguished Scientist Award.
The award and honorarium will be presented to Dr. Skolnick at a reception and dinner in Morgantown on March 18.

Super PSiFR: an automated pipeline for protein structure prediction, function annotation, and virtual screening.
In the post-genomic era, the annotation of protein function facilitates the understanding of various biological processes. Over the past decade, we have developed a suite of methods for predicting protein tertiary structure and function, ligand-binding sites, protein-protein interactions, protein-DNA interactions, virtual ligand screening, drug targets of a given compound, as well as tools for the large-scale comparison of protein structures, interfaces, or pockets among all known, experimentally determined structures deposited in the Protein Data Bank. To serve the scientific community, we announce a unified, intuitive, web-based server, Super PSiFR, which combines all these tools that we have developed. In the first beta-release, Super PSiFR provides working modules for protein structure prediction, small-molecule binding prediction, DNA-binding predictions, enzymatic function predictions, and virtual screening. To try it out, simply upload a sequence, a structure, or a small-molecule compound, and select desired modules.
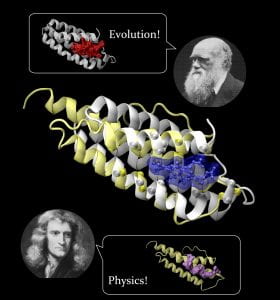
Study Suggests Drug Side Effects Inevitable; Basic Physics Enabled Early Biochemistry
A new study of both computer-created and natural proteins suggests that the number of unique pockets – sites where small molecule pharmaceutical compounds can bind to proteins – is surprisingly small, meaning drug side effects may be impossible to avoid. The study also found that the fundamental biochemical processes needed for life could have been enabled by the simple physics of protein folding.
Studying a set of artificial proteins and comparing them to natural proteins, researchers at the Georgia Institute of Technology have concluded that there may be no more than about 500 unique protein pocket configurations that serve as binding sites for small molecule ligands. Therefore, the likelihood that a molecule intended for one protein target will also bind with an unintended target is significant, said Jeffrey Skolnick, a professor in the School of Biology at Georgia Tech. Read more…

“Why not consider a spherical protein? Implications for backbone hydrogen bonding for protein structure and function.”
INSIDE COVER: Physical Chemistry Chemical Physics.
Abstract: The intrinsic ability of protein structures to exhibit the geometric features required for molecular function in the absence of evolution is examined in the context of three systems: the reference set of real, single domain protein structures, a library of computationally generated, compact homopolypeptides, artificial structures with protein-like secondary structural elements, and quasi-spherical random proteins packed at the same density as proteins but lacking backbone secondary structure and hydrogen bonding. Without any evolutionary selection, the library of artificial structures has similar backbone hydrogen bonding, global shape, surface to volume ratio and statistically significant structural matches to real protein global structures. Moreover, these artificial structures have native like ligand binding cavities, and a tiny subset has interfacial geometries consistent with native-like protein–protein interactions and DNA binding. In contrast, the quasi-spherical random proteins, being devoid of secondary structure, have a lower surface to volume ratio and lack ligand binding pockets and intermolecular interaction interfaces. Surprisingly, these quasi-spherical random proteins exhibit protein like distributions of virtual bond angles and almost all have a statistically significant structural match to real protein structures. This implies that it is local chain stiffness, even without backbone hydrogen bonding, and compactness that give rise to the likely completeness of the library solved single domain protein structures. These studies also suggest that the packing of secondary structural elements generates the requisite geometry for intermolecular binding. Thus, backbone hydrogen bonding plays an important role not only in protein structure but also in protein function. Such ability to bind biological molecules is an inherent feature of protein structure; if combined with appropriate protein sequences, it could provide the non-zero background probability for low-level function that evolution requires for selection to occur.

New Study Classifies and Analyzes Protein-Protein Interfaces
Interactions between proteins are at the heart of cellular processes, and those interactions depend on the interfaces where the direct physical contact occurs. A new study published this week suggests that there may be roughly a thousand structurally-distinct protein-protein interfaces — and that their structures depend largely on the simple physics of the proteins.

Super PSiFR: an automated pipeline for protein structure prediction, function annotation, and virtual screening.
In the post-genomic era, the annotation of protein function facilitates the understanding of various biological processes. Over the past decade, we have developed a suite of methods for predicting protein tertiary structure and function, ligand-binding sites, protein-protein interactions, protein-DNA interactions, virtual ligand screening, drug targets of a given compound, as well as tools for the large-scale comparison of protein structures, interfaces, or pockets among all known, experimentally determined structures deposited in the Protein Data Bank. To serve the scientific community, we announce a unified, intuitive, web-based server, Super PSiFR, which combines all these tools that we have developed. In the first beta-release, Super PSiFR provides working modules for protein structure prediction, small-molecule binding prediction, DNA-binding predictions, enzymatic function predictions, and virtual screening. To try it out, simply upload a sequence, a structure, or a small-molecule compound, and select desired modules.
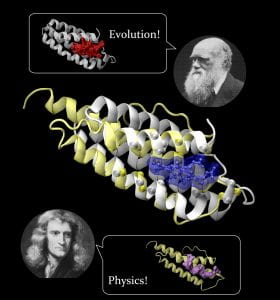
Study Suggests Drug Side Effects Inevitable; Basic Physics Enabled Early Biochemistry
A new study of both computer-created and natural proteins suggests that the number of unique pockets – sites where small molecule pharmaceutical compounds can bind to proteins – is surprisingly small, meaning drug side effects may be impossible to avoid. The study also found that the fundamental biochemical processes needed for life could have been enabled by the simple physics of protein folding.
Studying a set of artificial proteins and comparing them to natural proteins, researchers at the Georgia Institute of Technology have concluded that there may be no more than about 500 unique protein pocket configurations that serve as binding sites for small molecule ligands. Therefore, the likelihood that a molecule intended for one protein target will also bind with an unintended target is significant, said Jeffrey Skolnick, a professor in the School of Biology at Georgia Tech. Read more…

“Why not consider a spherical protein? Implications for backbone hydrogen bonding for protein structure and function.”
INSIDE COVER: Physical Chemistry Chemical Physics.
Abstract: The intrinsic ability of protein structures to exhibit the geometric features required for molecular function in the absence of evolution is examined in the context of three systems: the reference set of real, single domain protein structures, a library of computationally generated, compact homopolypeptides, artificial structures with protein-like secondary structural elements, and quasi-spherical random proteins packed at the same density as proteins but lacking backbone secondary structure and hydrogen bonding. Without any evolutionary selection, the library of artificial structures has similar backbone hydrogen bonding, global shape, surface to volume ratio and statistically significant structural matches to real protein global structures. Moreover, these artificial structures have native like ligand binding cavities, and a tiny subset has interfacial geometries consistent with native-like protein–protein interactions and DNA binding. In contrast, the quasi-spherical random proteins, being devoid of secondary structure, have a lower surface to volume ratio and lack ligand binding pockets and intermolecular interaction interfaces. Surprisingly, these quasi-spherical random proteins exhibit protein like distributions of virtual bond angles and almost all have a statistically significant structural match to real protein structures. This implies that it is local chain stiffness, even without backbone hydrogen bonding, and compactness that give rise to the likely completeness of the library solved single domain protein structures. These studies also suggest that the packing of secondary structural elements generates the requisite geometry for intermolecular binding. Thus, backbone hydrogen bonding plays an important role not only in protein structure but also in protein function. Such ability to bind biological molecules is an inherent feature of protein structure; if combined with appropriate protein sequences, it could provide the non-zero background probability for low-level function that evolution requires for selection to occur.

New Study Classifies and Analyzes Protein-Protein Interfaces
Interactions between proteins are at the heart of cellular processes, and those interactions depend on the interfaces where the direct physical contact occurs. A new study published this week suggests that there may be roughly a thousand structurally-distinct protein-protein interfaces — and that their structures depend largely on the simple physics of the proteins.

Factors Beyond Crowding
Using large-scale computer simulations, researchers at the Georgia Institute of Technology have identified the most important factors affecting how molecules move through the crowded environment inside living cells.
